
| AG | Intensivmedizin |
Leiter

Forschungsschwerpunkt: |
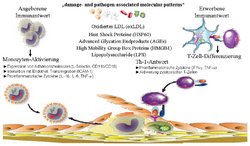
Akute Koronarsyndrome (ACS) werden durch eine Ruptur atherosklerotischer Plaques gefolgt von einer koronaren Thrombose verursacht. Die Ursachen für eine Plaqueinstabilität sind bisher unzureichend verstanden. In der Schulterregion der Plaque, also der Stelle mit dem größten Rupturrisiko, lassen sich vor allem Makrophagen und T-Lymphozyten nachweisen. Nicht nur im Bereich dieser „culprit lesion“, sondern auch systemisch lassen sich gesteigerte inflammatorische Prozesse im Sinne einer Aktivierung neutrophiler Granulozyten, Monozyten / Makrophagen und T-Lymphozyten nachweisen.
Das angeborene Immunsystem wird dabei über sog. „pathogen recognition receptors“ (PRRs) aktiviert. Hierzu zählen zellassoziierte Rezeptoren, wie die Toll-like-Rezeptoren (TLRs), der Rezeptor für Advanced Glycation Endproducts (RAGE), die Scavenger-Rezeptoren, und die lösliche Rezeptoren, wie z. B. LPS bindendes Protein (LBP), Komplementfaktoren und der lösliche Rezeptor für AGEs (s-RAGE). Als mögliche exogene und endogene Liganden für diese PRRs werden sog. „pathogen-associated“ (PAMPs) oder „damage-associated molecular patterns“ (DAMPs) diskutiert.
Reichen die angeborenen Immunreaktionen nicht aus, durchbrechen also PAMPs oder DAMPs die ersten Abwehrlinien, werden erworbene Immunreaktionen ausgelöst und Lymphozyten aktiviert. Extrazelluläre Antigene werden von antigenpräsentierenden Zellen (z.B. dendritische Zellen) aufgenommen, prozessiert und zusammen mit dem oberflächengebundenen MHC-2 Molekül naiven CD4+ T-Zellen präsentiert. Der T-Zell-Rezeptor (TCR) der CD4+ T-Zellen bindet an den Antigen-MHC-2-Komplex der antigenpräsentierenden Zelle. In einer aktivierten T-Zelle kommt es über unterschiedliche Signaltransduktionswege zu einer Aktivierung der Transkriptionsfaktoren NFAT, AP-1 und NF-kB. Das Verhältnis dieser Transkriptionsfaktoren zueinander entscheidet über die weitere Differenzierung der T-Zelle und ihre Zytokin-Produktion. Aus atherosklerotischen Läsionen gewonnene T-Zellklone sezernieren überwiegend Th1-zellspezifische Zytokine (IFN-g, TNF-a), während die Th2-spezifischen Zytokine (IL-4, IL-5) nur eine untergeordnete Rolle spielen. Auch im peripheren Blut von Patienten mit akutem Koronarsyndrom zeigen zirkulierende CD4+ T-Lymphozyten eine gesteigerte IFN-g- und TNF-a-Produktion. Sowohl im peripheren Blut als auch in der Schulterregion der Plaque lässt sich eine spezifische T-Lymphozyten-Subpopulation nachweisen, welche die Expression des co-stimulatorischen Rezeptors CD28 verloren hat. Diese CD4+CD28null T-Zellen sind in der Lage exzessive Mengen von IFN-g und zytolytischen Proteinen freizusetzen. Neben anderen proinflammatorischen Mechanismen werden diese T-Zellen mit atheromatöser Plaque-Destabilisierung und gesteigertem endothelialen Zelltod bei Patienten mit akuten Koronarsyndromen in Verbindung gebracht.
Ziel des Forschungsprojektes ist es, bei Patienten mit ACS die Reaktionen des angeborenen und erworbenen Immunsystems näher zu charakterisieren, mögliche Mechanismen einer überschießenden Aktivierung zu identifizieren und etwaige therapeutische Ansätze zu prüfen, welche die Immunantwort modulieren könnten.
Forschungsschwerpunkt: |
Bis zu 10 % aller Patienten mit akutem Koronarsyndrom (ACS) entwickeln einen kardiogenen Schock, welcher trotz rascher Wiederherstellung eines effektiven koronaren Blutflusses (Akut-Koronarintervention, Fibrinolyse, Antikoagulation und Thrombozytenaggregationshemmung) mit einer unverändert hohen Mortalität von 60-70 % assoziiert ist. Ein Patient im infarktbedingten kardiogenen Schock verstirbt nach heutigem pathophysiologischen Verständnis nicht an einer verschlossenen Koronararterie, sondern an den Folgen einer sich entwickelnden systemischen Inflammationsreaktion (SIRS), an dessen Ende ein Multiorganversagen steht. Durch Ischämie und Reperfusion sowie schockbedingte Organminderperfusionen werden endogene - „damage associated“ - und exogene - „pathogen associated“ - Liganden freigesetzt, welche eine angeborene und erworbene Immunantwort induzieren. Diese zunächst protektiv wirkende Immunreaktion kann aus bisher nicht geklärten Gründen in ein überschießendes SIRS und ein Multiorgandysfunktionssyndrom mit schlechter Prognose übergehen.
Ziel des Forschungsprojektes ist es, bei Patienten mit infarktbedingtem kardiogenen Schock die Reaktionen des angeborenen und erworbenen Immunsystems näher zu charakterisieren, mögliche Mechanismen einer überschießenden Aktivierung zu identifizieren und etwaige therapeutische Ansätze zu prüfen, welche die Immunantwort modulieren könnten.
Forschungsschwerpunkt: |
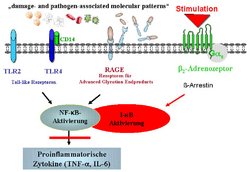
Im manifesten septischen und kardiogenen Schock lassen sich viele proinflammatorische Signalwege nicht mehr hemmen. Seit langen ist bekannt, dass eine ß2-Adrenozeptor-Stimulation in Monozyten eine LPS-vermittelte proinflammatorische Zytokin-Antwort über eine cAMP / ß-Arrestin-vermittelte I-kB-Aktivierung hemmen kann, den sog. adrenergen antiinflammatorischen Signalweg. Exogene und endogene Liganden (PAMPs und DAMPs) stimulieren „pathogen recognition receptors“ – TLR2, TLR4, RAGE – und induzieren eine NF-κB-vermittelte proinflammatorische Immunantwort. Eine gleichzeitige ß2-adrenerge Stimulation steigert die Expression von I-κB und blockiert dadurch die NF-κB-vermittelte Zytokinfreisetzung.
Im septischen Schock kommt es häufig infolge einer endogenen oder pharmakologischen Katecholaminwirkung zu einer „down“-Regulation monozytärer ß2-Adrenozeptoren (ß2-AR). Durch diese ß2-AR- „down“-Regulation kann die cAMP-vermittelte Hemmung der Freisetzung proinflammatorischer Zytokine nicht mehr aufrechterhalten werden. Eine proinflammatorische monozytäre Zytokinfreisetzung ist die Folge. Auch im kardiogenen Schock kommt es zu einer „down“-Regulation von ß-AR. Dies ist bisher für ß1-AR auf Kardiomyozyten bekannt. Ob auch die monozytäre ß2-AR-Expression im kardiogenen Schock einer katecholaminbedingten „down“-Regulation unterliegt, wurde bisher noch nicht untersucht.
Ziel des Forschungsprojektes ist es, bei Patienten im septischen / kardiogenen Schock den adrenergen antiinflammatorische Signalweg zu untersuchen: Welche Signalwege bedingen diese Dysregulation im septschen / kardiogenen Schock? Welchen Einfluß haben exogen zugeführte Katecholamine? Gibt es Unterschiede innerhalb der Katecholamine (z.B. Dobutamin vs. Noradrenalin)? Gibt es Möglichkeiten für eine therapeutische Intervention?
Forschungsschwerpunkt: |
Unfraktioniertes Heparin (UFH) ist bei kritisch kranken Patienten ein häufig genutztes Antikoagulanz zur Prophylaxe und Therapie von Thrombosen und zur Antikoagulation bei extrakorporalen Verfahren (Dialyse, Herzlungenmaschine). Durch die extrakorporale Zirkulation werden auf zellulärer und humoraler Ebene folgende Systeme aktiviert: Thrombozyten, Leukozyten, Komplementsystem, Gerinnungskaskade. Dies führt zu einer gesteigerten Thrombozytenaktivierung, thrombo-embolischen Ereignissen und einer Abnahme der Thrombozytenzahl, die für die Fortführung der Dialysebehandlung oftmals limitierend ist. Ferner ist in bis zu 3% der Fälle mit dem Auftreten einer Heparin-induzierten Thrombozytopenie Typ II (HIT-II) zu rechnen. Die Inzidenz einer HIT-II beträgt bei internischen Patienten 0,1 – 2,5%, bei orthopädischen und herzchirurgischen Eingriffen bis zu 2 – 3%.
Folgende Alternativ-Antikoagulanzien werden bei kritisch Kranken mit der Notwendigkeit eines extrakorporalen Dialyseverfahrens untersucht:
- der GPIIb/IIIa-Rezeptorantagonist - Tirofiban
- der direkter Thrombininhibitor - Argatroban
- Citrat als extrakorporales Antikoagulanz
Publikationen |
- Originalarbeiten
- Link A, Hummel B, Schwerdt H, Schwamborn J, Jung F, Schieffer H. Influence of neutrophil separation on the expression of adhesion molecules. Clin Hemorheol Microcirc 1997; 117: 175-180.
- Link A, Schwerdt H, Hennen B, Böhm M. Polymorphonuclear neutrophils in myocardial ischemia and reperfusion injury - Influence of coronary intervention. ZKardiol 2004; 93: 605-611.
- Link A, Lenz M, Legner D, Böhm M, Nickenig G. Telmisartan inhibits ß2-integrin Mac-1 expression in human T-lymphocytes. J Hypertens 2006; 24: 1891-8.
- Link A, Ayadhi T, Böhm M, Nickenig G. Rapid immunomodulation by rosuvastatin in patients with acute coronary syndrome. Eur Heart J 2006; 27: 2945-55.
- Link A, Girndt M, Selejan S, Rbah R, Böhm M. Tirofiban preserves platelet loss during continuous rnal replacement therapy in a randomised prospective open-blindet pilot study. Crit Care 2008; 12 R111
- Link A, Selejan S, Maack C, Lenz M, Böhm M. Phosphodiesterase 4 inhibition but not beta-adrenergic stimulation suppresses tumor necrosis factor-alpha release in peripheral blood mononuclear cells in septic shock. Crit Care 2008: 12: R159
- Link A, Girndt M, Selejan S, Mathes A, Böhm M, Rensing H. Argatroban for anticoagulation in continuous renal replacement therapy. Crit Care Med 2009; 37: 105-10
- Selejan S, Hewera L, Walter F, Böhm M, Link A. sRAGE influences mortality in cardiogenic shock. Clin Res Cardiol 2009;98:P 1389.

