
Gliome
Allgemeines
Zu den Gliomen gehören die hirneigenen Tumoren, die aus den Gliazellen entstehen. Diese stellen das Stütz- und Nährgewebe der Nervenzellen dar. Je nachdem von welchen Zellen der Tumor ausgeht, unterscheidet man verschiedene Gliome, wie z.B. Astrozytome, Oligodendrogliome, Ependymone oder auch Mischformen wie Oligoastrozytome. Gliome machen etwa 45-50% der intrakraniellen Tumore aus. Etwa 50% der Gliome sind Glioblastome (Astrozytome WHO Grad IV). Astrozytome (WHO Grad I-III) machen etwa 25% der Gliome aus und Oligodendrogliome weniger als 5-18%. Ependymome werden mit einer Häufigkeit von 2-9% angegeben. Gliome können in allen Arealen des Gehirns auftreten und in seltenen Fällen auch im Rückenmark oder der Augenhöhle.
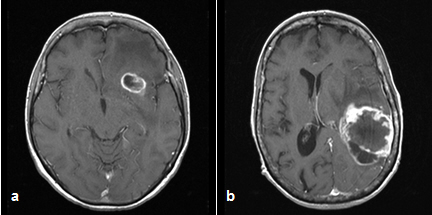
Darstellung von Glioblastomen im MRT an verschiedenen Lokalisationen:
frontal links (a) und temporoparietal links (b)
Klassifikation
Wie es für Hirntumore üblich ist, werden die Gliome nach der WHO-Klassifikation eingeteilt, welche sich nach dem histologischen Befund, dem Wachstumsverhalten, der Rezidivwahrscheinlichkeit und somit auch der Prognose richtet.
- WHO Grad I: Tumor ist histologisch gutartig und durch vollständige Resektion in der Regel heilbar, Rezidiv unwahrscheinlich (z.B. pilozytisches Astrozytom, Ependymom)
- WHO Grad II: Tumor ist histologisch gutartig, jedoch infiltratives Wachstum und Rezidivneigung, Überleben nicht wesentlich eingeschränkt (z.B. diffuses Astrozytom, langsam wachsendes Oligodendrogliom und Oligoastrozytom)
- WHO Grad III: Tumor ist histologisch bösartig, Überlebenszeit ist eingeschränkt (z.B. anaplastisches Astrozytom, anaplastisches Ependymom, anaplastisches Oligodendrogliom, anaplastisches Oligoastrozytom)
- WHO Grad IV: Tumor ist äußerst bösartig, aggressives Wachstum, Überlebenszeit deutlich reduziert (z.B. Glioblastom)
Die Symptome hängen wesentlich von der Lokalisation der Tumoren ab und von ihrer Wachstumsgeschwindigkeit. Es können Hirndruckzeichen (z.B. Kopfschmerzen, Müdigkeit, Übelkeit, Schwindel), neurologische Ausfälle (z.B. Lähmungen, Sehstörungen, Gehstörungen, Sprachstörungen) oder epileptische Anfälle auftreten. Zudem kann es zu psychischen Veränderungen wie Desorientiertheit oder leichter Reizbarkeit kommen.
Die Diagnostik erfolgt durch CT- und MRT-Bildgebungen. Diese Bilder können auch während der Operation zur Neuronavigation benutzt werden, um den Tumor und umliegende Hirnstrukturen genau lokalisieren zu können. Differenzialdiagnostisch können auch nuklearmedizinische Untersuchungen wie die PET (Positronen-Emissions-Tomographie) oder SPECT (Einzelphotonen-Emissionscomputertomographie) zur Anwendung kommen. Je nach Lokalisation können auch noch weitere präoperative Untersuchungen, wie z.B. die navigierte transkranielle Magnetstimulation (nTMS) oder das Diffusions-Tensor-Imaging Fibretracking (Traktographie) sinnvoll sein.
Die Therapie richtet sich nach der Art und Lokalisation des Tumors sowie dem Alter und Allgemeinbefinden des Patienten und wird für jeden Patienten individuell und in interdisziplinärer Zusammenarbeit mit anderen beteiligten Fachrichtungen wie zum Beispiel der Klinik für Strahlentherapie oder der Klinik für Innere Medizin I festgelegt und mit dem Patienten abgestimmt. Im Vordergrund der neurochirurgischen Behandlung steht meist die operative Entfernung des Tumors. Diese erfolgt in mikrochirurgischer Technik und in der Regel mit Hilfe der Neuronavigation und verschiedener Techniken des intraoperativen Monitorings wie elektrophysiologische Untersuchungen, Fluoreszenzdarstellung des Tumorgewebes mit 5-ALA, Wachoperationen sowie intraoperative CT- oder Ultraschallbildgebung. Im einigen Fällen kann vor einer Tumorentfernung auch eine stereotaktische Biopsie zur Diagnosesicherung sinnvoll sein.
Je nach Tumorart und Resektionsausmaß kann sich an eine operative Entfernung eine Nachbehandlung anschließen. Im Falle eines Glioblastoms z.B. bedeutet das eine anschließende kombinierte Strahlen- und Chemotherapie. Bei Rezidiven kann intraoperativ zusätzlich ein lokalwirksames Chemotherapeutikum in die Resektionshöhle eingebracht werden.
Molekularbiologische Untersuchungen des Tumorgewebes können bei bestimmten Tumorarten voraussagen, wie gut die unterschiedlichen Therapien wirken können. Durch sie lässt sich die Therapie noch genauer und individueller planen. Hierin liegt auch ein wesentlicher Schwerpunkt unserer neuroonkologischen Forschung.
Unsere Expertise
- Komplettes Spektrum der Neuroonkologie
- Enge interdisziplinäre Zusammenarbeit mit der Klinik für Strahlentherapie, Klinik für Innere Medizin I etc.
- Teilnahme am onkologischenTumor-board
- Neuroonkologische Forschung
Ärztlicher Ansprechpartner
OA Prof Dr. med. R. Ketter
E-Mail: Ralf.Ketter @uks.eu
HSA-Sprechstunde:
Di 09:00-14:00
Anmeldung
HSA-Sprechstunde:
Tel.: 06841-1624412

