
Entwicklungswege bei Gliomen
veröffentlicht in Medizinische Genetik 2002, Bd. 14 von Prof. Dr. Klaus Zang
Unter Hirntumoren werden alle innerhalb des Schädels liegenden Geschwülste des Zentralnervensystems zusammengefasst. Sie zeigen eine enorme histologische und zytologische Variationsbreite und eine erhebliche intratumorale Heterogenität. Die WHO-Klassifikation differenziert mehr als 120 Subtypen, deren Beschreibung von Zellen des embryonalen und adulten Gehirns abgeleitet ist. Dem entspricht bereits nach dem heutigen Kenntnisstand eine Vielzahl teils übereinstimmender, teils sich ausschließender primärer und progressions-assoziierter somatischer genetischer Veränderungen, deren Nachweis sowohl differentialdiagnostisch, als auch prognostisch hilfreich sein kann.
Hirntumoren sind vergleichsweise selten. Sie liegen in Deutschland bei den Frauen mit 3,7 % auf dem 10. Platz und bei den Männern mit 2,7 % auf dem 14. Platz der gesamten Tumorinzidenz. Internationale Zahlen sind vergleichbar. Unter der unscharfen klinischen Bezeichnung "Hirntumoren" werden üblicherweise alle intrakraniellen Geschwülste unterschiedlicher histogenetischer Entwicklung zusammengefasst. Nur etwa 50 % sind neuroepitheliale Tumoren, unter denen wiederum neuronale Tumoren nur eine verschwindende Minderheit ausmachen. Die überwiegende Mehrzahl sind Gliome. Weitere rund 25 % sind Meningeome, obwohl die Hirnhäute weniger als 5 % der intrakraniellen Masse ausmachen. Unter den restlichen 25 % finden sich etwa zu gleichen Teilen Geschwülste der Hirnnerven, vor allem Schwannome, Tumoren der Sellaregion, d.h. Hypophysenadenome und Kraniopharyngeome, Gefäß- oder Fehlentwicklungstumoren sowie schwer zu klassifizierende (Misch-)Tumoren. Hirntumoren treten in der Regel sporadisch auf; sie können jedoch auch Teil eines autosomal-dominanten (Tumor-) Syndroms sein und stehen dann meist in ätiologischem Zusammenhang mit der Keimbahn- oder Keimzellmutation eines Tumor-Suppressorgens.
Die histologisch-zytologische Variationsbreite der Hirntumoren ist ganz erheblich. Sie liegt fast in der Größenordnung aller übrigen Körpertumoren zusammen. Einige der genetischen Veränderungen können bei fast allen Hirntumoren auftreten. Andere sind spezifisch für bestimmte Tumorentitäten. Die phänotypischen Unterschiede der einzelnen Hirntumoren beruhen offenbar zusätzlich auf unterschiedlichen Genexpressionsmustern der betreffenden Matrixzellen, aber auch auf unterschiedlichen überwiegend noch nicht erforschten zusätzlichen genetischen Veränderungen. Im Rahmen dieser kurzen Übersicht kann nur auf die häufigsten Tumoren und auf deren besondere biologische Eigentümlichkeiten eingegangen werden.
Dazu gehört einmal die anatomische Situation der geschlossenen Schädelhöhle, die keine Volumenzunahme zulässt, sondern bei Entwicklung eines Tumors Volumenverschiebungen und eine Druckzunahme auslöst, die unabhängig von der Bösartigkeit des Tumors letztlich zum Tode führt. Eine extracerebrale Metastasierung erfolgt fast nie. Die Forschung über neuronale Stammzellen hat neue Erkenntnisse bezüglich des ganz ungewöhnlichen migratorischen Verhaltens von Gliomen und der Häufigkeit von Hirntumoren mit embryonalen Charakteristika geliefert. Histologisch wird auch bei den intrakraniellen Tumoren üblicherweise zwischen benigne und maligne unterschieden. Wie bei den Gliomen zu zeigen sein wird, ist eine solche Unterscheidung jedoch nur von untergeordneter, überwiegend auf die Überlebenszeit reduzierter, Bedeutung. Gliome sind kaum kurativ therapierbar. Mit Ausnahme jugendlicher pilozytischer Astrozytome, einiger niedriggradiger Oligodendrogliome und der Mehrzahl der Meningeome gelingt die operative Entfernung aller Tumorzellen selten. Die Tumoren rezidivieren, häufig in malignerer Form. Die in ihrer Dignität und relativen Häufigkeit deutlich abweichenden kindlichen Hirntumoren sind nicht Gegenstand dieser Übersicht.
Gliome
Diese Tumoren werden üblicherweise histologisch und nach ihrer Dignität, d.h. ihrem Differenzierungsgrad (Grad I-IV), entsprechend einer WHO-Klassifikation eingeteilt (Kleihues and Cavenee, 2000). Von den neuroepithelialen Tumoren sind rund 90 % Gliome, davon 70 % Astrozytome, 7 % Oligodendrogliome, bzw. oligo-astrozytäre Mischtumoren und 3 % Ependymome unterschiedlicher Dignität. Alle übrigen sind beim Erwachsenen selten. Astrozytäre Tumoren werden eingeteilt in, meist juvenile, pilozytische Astrozytome (Grad I, meist auch nicht höhergradig rezidivierend), diffuse Astrozytome (Grad II), anaplastische Astrozytome (Grad III) und Glioblastome (Grad IV). Hinzu kommen die deutlich selteneren Oligodendrogliome und Oligoastrozytome (Grad II, bzw. III), sowie Ependymome (Grad II, bzw. III). Fast die Hälfte der Gliome sind Glioblastome, die wohl bösartigsten und therapieresistentesten Tumoren des Menschen überhaupt.
Die Bezeichnung ?diffuse? Gliome weist auf die ungewöhnliche Fähigkeit auch niedriggradiger Gliome hin, das umliegende Hirngewebe weiträumig zu infiltrieren, ohne zunächst dessen Zyto- und Faserarchitektur und neuronale Funktionen zu zerstören. Dieses Fehlen klarer Tumorbegrenzungen ist (neben der weitgehenden Chemo- und Strahlenresistenz) der Grund für die Unmöglichkeit auch niedriggradiger Hirntumoren operativ restlos zu entfernen. Die Gradierung selbst folgt einfachen histologischen Kriterien: Bereits niedriggradiger Gliome zeigen eine erhebliche zytologische Polymorphie; anaplastische Gliome weisen eine zunehmende mitotische Aktivität und zunehmende Zelldichte auf; Glioblastome sind zusätzlich außer durch weiter zunehmende Proliferationsaktivität durch eine starke Angiogenese und durch Mikronekrosen gekennzeichnet. Sowohl die histologische als auch die genetische Charakterisierung von Gliomen wird durch die oft erhebliche intratumorale Heterogenität erschwert. Molekulargenetische (LOH-) Ergebnisse am Tumorhomogenat können quantitativ und im Verteilungsmuster von immunzytochemischen und FISH-Ergebnissen am Schnittpräparat und diese wiederum von CGH-Ergebnissen nach DOP-PCR mikrodissezierter kleiner Areale erheblich abweichen (Jung et al. 1999). Die Ergebnisse der verschiedenen Arbeitsgruppen und methodischen Richtungen stimmen überein bezüglich der Zunahme typischer genetischer Veränderungen mit zunehmender Malignität, sowie bezüglich eines einheitlichen Grundgerüsts sich summierender genetischer Veränderungen (Kleihues and Cavenee, 2000; Rasheed et al. 1999). Überraschenderweise kommt dabei einer zunehmenden numerischen Chromosomeninstabilität mehr unter Verlust, als unter Zugewinn, ganzer Chromosomen(-arme), bzw. größerer mikroskopisch noch auflösbarer Chromatinstrukturen eine erhebliche Bedeutung zu (Übersicht bei Kleihues and Cavenee, 2000; Loeper et al. 2001). Die Aktivierung bekannter, aber auch noch unbekannter Onkogene scheint nach den bisherigen Erkenntnissen eine wesentliche Rolle zu spielen. Es finden sich mit zunehmender Malignität einige charakteristische Amplifikationseinheiten unter Einbeziehung bekannter Wachstumsfaktor-Liganden und -Rezeptoren. Es sind erst wenige beteiligte Tumorsuppressorgene definiert. Die häufigsten numerischen Veränderungen sind +7, -10 und ?22, die häufigsten strukturellen Veränderungen sind 9p-(CDKN2A(P16)/ARF(P14)/CDKN2B(P15)), 10q-(PTEN) und 17p-(P53); die am häufigsten zu beobachtenden Amplifikationen liegen auf 7 (u. a. EGFR, selten CDK6) bzw. auf 12q (u. a. MDM2 oder CDK4).
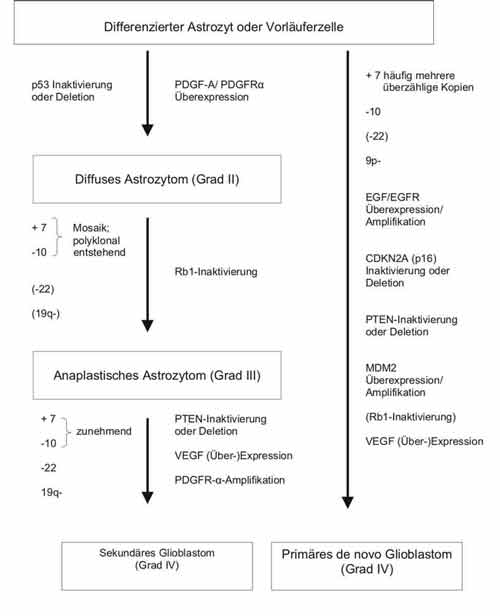
Pilozytische Astrozytome (Grad I) zeigen zytogenetisch, aber auch molekulargenetisch, nur wenige und überwiegend uneinheitliche Veränderungen. Seltene höhergradige (Rezidiv-) Tumoren zeigen astrozytomtypische Veränderungen. Diffuse Astrozytome (Grad II) sind durch p53-Mutationen bzw. -Deletionen bei sonst ebenfalls seltenen und überwiegend unsystematischen Veränderungen gekennzeichnet. Eine mit Hilfe von FISH, nicht jedoch in der Zellkultur nachgewiesene variable Mosaik-Trisomie 7 und Monosomie 10 wird als Progressionsparameter diskutiert. Das gleiche gilt für andere mit in situ-Methoden bei einem Teil der Fälle nachgewiesene fokale, für höher gradige Gliome typische Veränderungen. Dies gilt insbesondere für die Überexpression von PDGFa-Ligand und -Rezeptor im Sinne eines autokrinen Zyklus, die als Mitursache der hohen Migrationsfähigkeit dieser Tumorzellen diskutiert wird. Trotz der häufig nachgewiesenen p53-Verluste findet sich kein Anhalt für eine erhöhte Aneuploidie.
Anaplastische Astrozytome sind eher histologisch und durch eine quantitative Zunahme der genetischen Veränderungen, z.B. Allelverluste (LOH) auf 19q als durch weitere definierte qualitative Veränderungen von den niedriggradigen Astrozytomen abzugrenzen. Sie können fokal Glioblastom-spezifische Veränderungen zeigen. Bei rund 25 % der Fälle wird allerdings ein Verlust oder die Inaktivierung des Rb-Gens berichtet. Die Tumoren können sich nach einer Latenzzeit von oft mehreren Jahren primär oder als Rezidiv aus einem niedriggradigen diffusen Astrozytom entwickeln. Sie sind im Grunde ein Durchgangsstadium zum Glioblastom.
Glioblastome bestätigen in ihren genetischen Veränderungen die klinisch-epidemiologischen Befunde von zumindest zwei unterschiedlichen Subtypen. Einmal das weniger aggressive sekundäre Glioblastom jüngerer Menschen, das sich innerhalb von 5 bis 10 Jahren (meist als Rezidiv) aus einem niedriggradigen Astrozytom entwickelt und zum anderen das aggressivere primäre Glioblastom älterer Menschen (Median 55 Jahre). Nach dem jetzigen Kenntnisstand gibt es bei primären und sekundären Glioblastomen sowohl übereinstimmende genetische Veränderungen, wie Vermehrung der Kopienzahl von Chromosom 7, Verluste von Chromosom 10, bzw. homozygoter Aktivitätsverlust des PTEN-Gens und solche, die sich gegenseitig weitestgehend ausschließen. Dazu gehören die p53-Inaktivierung und PDGFA/PDGFRa-Überexpression beim sekundären und die p16-Inaktivierung und EGFR- und/oder MDM2-Amplifikation beim primären Glioblastom. Histologisch und in ihrer Therapieresistenz unterscheiden sich diese Tumoren nicht. Während das primäre Glioblastom in der Regel innerhalb von weniger als einem Jahr zum Tode führt, ist beim sekundären Glioblastom die durchschnittliche Lebenserwartung etwas länger. Neuere Ergebnisse deuten daraufhin, dass die starke Angiogenese korreliert ist mit einer EGF/EGFR-induzierten VEGF-Überexpression (Übersicht bei Maher et al. 2001).
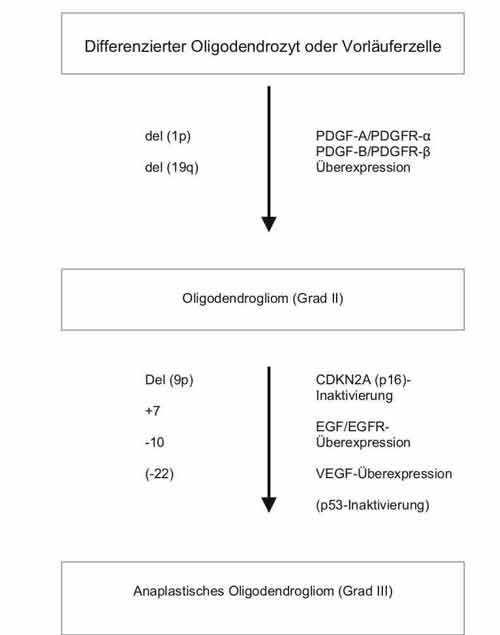
Oligodendrogliome haben in der Regel eine längere Anamnese und günstigere Prognose als Astrozytome. Sie infiltrieren geringer, sind dadurch besser operativ angehbar und lassen sich häufig chemotherapeutisch erfolgreich behandeln. Sie zeigen von Astrozytomen abweichende charakteristische genetische Veränderungen. Es sind meist nur molekulargenetisch und nicht zytogenetisch nachweisbare heterozygote Verluste der Chromosomenarme 1p und 19q, einzeln, meist jedoch in Kombination auftretend. Die entscheidenden Gene auf diesen Chromosomenarmen sind noch nicht bekannt, obwohl bereits sehr enge Konsensus-Regionen festgelegt sind. Im Zuge der Progression zu anaplastischen Tumoren entwickeln sich zusätzlich Astrozytom-spezifische Veränderungen, wie 9p-, und PDGFA/R- bzw. EGF/R-Überexpressionen.
Oligoastrozytome sind nicht allzu selten und von besonderem histogenetischem, wie molekulargenetischem Interesse. Bei ihnen stellt sich die Frage nach einem polyklonalen Mischtumor unbekannter (exogener) Induktion oder einer unterschiedlichen pathologischen Ausdifferenzierung und Proliferation einer gemeinsamen embryonalen (OIIA-) Vorläuferzelle. Die Tumoren zeigen sowohl Oligodendrogliom- als auch Astrozytom-typische genetische Veränderungen. Es ist noch nicht definitiv geklärt, ob die genetischen Veränderungen sich in den beiden Zelltypen unterscheiden. LOH-Untersuchungen am Homogenat können die Information nicht liefern; FISH-Untersuchungen am Schnittpräparat und CGH, bzw. LOH-Untersuchungen an Mikroklonen deuten auf unterschiedliche Verteilungsmuster hin (Kraus et al. 2001; eigene unpublizierte Befunde. Nienhaus, Weber et al. in Vorbereitung).
Möglicherweise können die neuronalen Stammzellbefunde der letzten Jahre sowohl zu diesem Phänomen als auch zu den migratorischen Fähigkeiten von Gliomzellen wichtige Informationen liefern. Auch im adulten Gehirn finden sich offenbar Lager neuronaler Stammzellen bevorzugt im subventrikularen Bereich und im Hippocampus, beides für Hirntumoren untypische Regionen. Es wird diskutiert, dass mutativ veränderte Stammzellen sowohl ihre embryonalen migratorischen Fähigkeiten wiedergewinnen und die typischen Bahnen einschlagen können, aber auch, dass sie sich dann an den Gliom-Prädilektionsorten vermehren und dort einen Tumor entwickeln (Übersicht bei Maher et al., 2001). Die Entstehung von Mischtumoren, wie Oligoastrozytomen, aber auch Gliosarkomen könnte auf diese Weise eine Erklärung finden, aber auch von nicht allzu selten auftretenden multiplen oder symmetrischen Schmetterlings-Gliomen, deren räumliche Distanz gegen eine intracerebrale Metastasierung spricht. Weder histologisch noch aufgrund der genetischen Veränderungen lässt sich zum gegenwärtigen Stand unserer Kenntnisse entscheiden, ob ein malignes Gliom durch Entdifferenzierung und Proliferation adulter Gliazellen oder durch Migration und Proliferation neuronaler Stammzellen entstanden ist.

